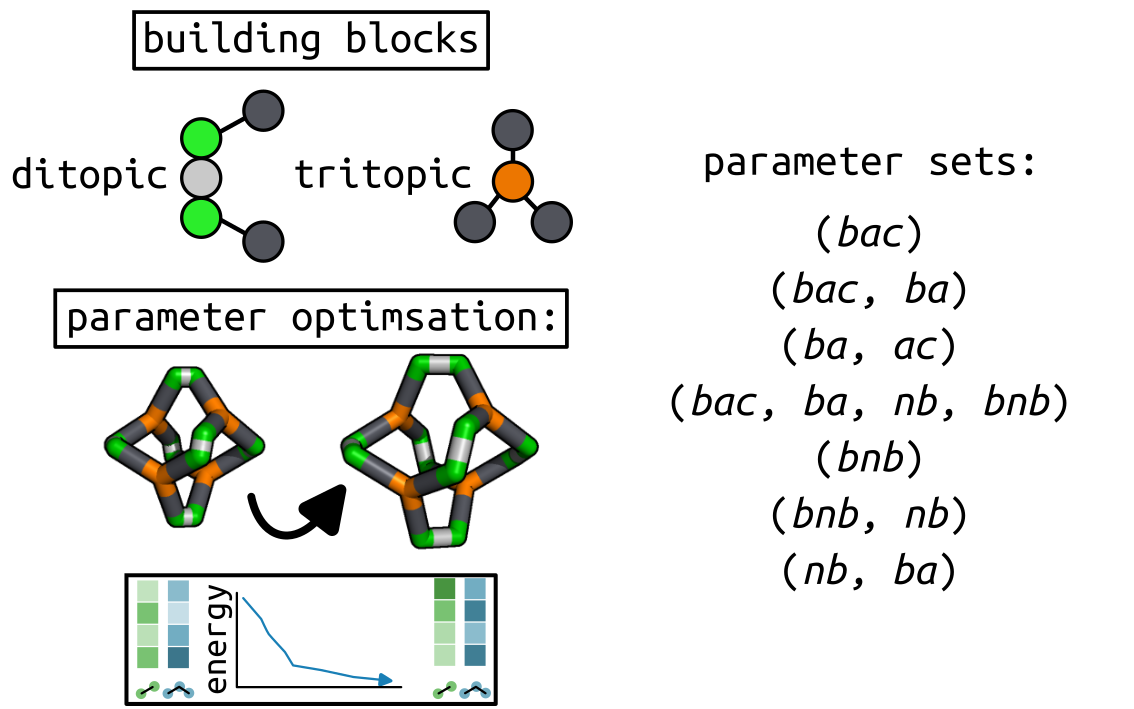
Target optimisation¶
Here I show the use of the cgexplore.scram.target optimisation() function
to optimise selected parameters of an input forcefield to minimise the energy
of a provided structure. We have a FourPlusSix cage, that is strained, with
building block parameters coming from the same building blocks in
recipe 4. We then apply a series of different forcefield
parameter optimisations.
We will start by defining our definer_dict for the constant terms, and the
associated bead library.
# Define a definer dictionary.
# These are constants, while different systems can override these
# parameters.
cg_scale = 2
constant_definer_dict = {
# Bonds.
"nb": ("bond", 1.0, 1e5),
# Angles.
"aca": ("angle", 180, 1e2),
"nba": ("angle", 180, 1e2),
# Nonbondeds.
"n": ("nb", 10.0, 1.0),
"a": ("nb", 10.0, 1.0),
"b": ("nb", 10.0, 1.0),
"c": ("nb", 10.0, 1.0),
}
# Define beads.
bead_library = cgx.molecular.BeadLibrary.from_bead_types(
# Type and coordination.
{"n": 3, "a": 2, "b": 2, "c": 2}
)
I skip this in the recipe, but we can use the same process as recipe 2 or recipe 5 to perform structure prediction. Here, we still define the building blocks and systems.
# Define your forcefield alterations as building blocks.
building_block_library = {
"ditopic": {
"precursor": cgx.molecular.TwoC1Arm(
bead=bead_library.get_from_type("c"),
abead1=bead_library.get_from_type("a"),
),
"mod_definer_dict": {
"ba": ("bond", 1.5 / cg_scale, 1e5),
"ac": ("bond", 1.5 / 2 / cg_scale, 1e5),
"bac": ("angle", 115, 1e2),
},
},
"tritopic": {
"precursor": cgx.molecular.ThreeC1Arm(
bead=bead_library.get_from_type("n"),
abead1=bead_library.get_from_type("b"),
),
"mod_definer_dict": {
"nb": ("bond", 3.0 / cg_scale, 1e5),
"bnb": ("angle", 120, 1e2),
},
},
}
# Define systems to predict the structure of.
systems = {
"cc3": {
"stoichiometry_map": {"tritopic": 2, "ditopic": 3},
"multipliers": (2,),
"vdw_cutoff": 2,
},
}
And the parameters we want to scan in different tests.
# Define a series of parameter explorations.
parameter_sets = [
("bac",),
("bac", "ba"),
("ba", "ac"),
("bac", "ba", "nb", "bnb"),
("bnb",),
("bnb", "nb"),
("nb", "ba"),
]
Time to iterate! Again, skipping the structure prediction, and just assuming we
have a target structure with a key in a database generated during structure
prediction. Once defined, we can run
cgexplore.scram.target_optimisation() over the parameters we are
interested in.
for system_name, syst_d in systems.items():
logger.info("doing system: %s", system_name)
# Merge constant dict with modifications from different systems.
merged_definer_dicts = cgx.systems_optimisation.merge_definer_dicts(
original_definer_dict=constant_definer_dict,
new_definer_dicts=[
building_block_library[i]["mod_definer_dict"]
for i in syst_d["stoichiometry_map"]
],
)
forcefield = cgx.systems_optimisation.get_forcefield_from_dict(
identifier=f"{system_name}ff",
prefix=f"{system_name}ff",
vdw_bond_cutoff=syst_d["vdw_cutoff"],
present_beads=bead_library.get_present_beads(),
definer_dict=merged_definer_dicts,
)
# A structure i have predicted earlier (using the same approach as
# recipe 2/5).
chosen_name = "cc3_2_4"
conformer_db_path = calc_dir / f"{chosen_name}.db"
conformer_db = cgx.utilities.AtomliteDatabase(conformer_db_path)
min_energy_structure = None
min_energy = float("inf")
for entry in conformer_db.get_entries():
if entry.properties["energy_per_bb"] < min_energy:
min_energy = entry.properties["energy_per_bb"]
min_energy_structure = conformer_db.get_molecule(
key=entry.key
)
num_bbs = entry.properties["num_bbs"]
for ps, parameters in enumerate(parameter_sets):
logger.info("doing %s", parameters)
database_path = data_dir / f"set_{ps}.db"
ffoptcalculation_dir = calc_dir / f"set_{ps}"
ffoptcalculation_dir.mkdir(exist_ok=True)
# To database.
cgx.utilities.AtomliteDatabase(database_path).add_molecule(
key=chosen_name,
molecule=min_energy_structure,
)
cgx.utilities.AtomliteDatabase(database_path).add_properties(
key=chosen_name,
property_dict={
"energy_per_bb": min_energy,
"num_bbs": num_bbs,
"forcefield_dict": (
forcefield.get_forcefield_dictionary()
),
},
)
cgx.scram.target_optimisation(
database_path=database_path,
target_key=chosen_name,
calculation_dir=ffoptcalculation_dir,
definer_dict=merged_definer_dicts,
modifiable_terms=parameters,
forcefield=forcefield,
)
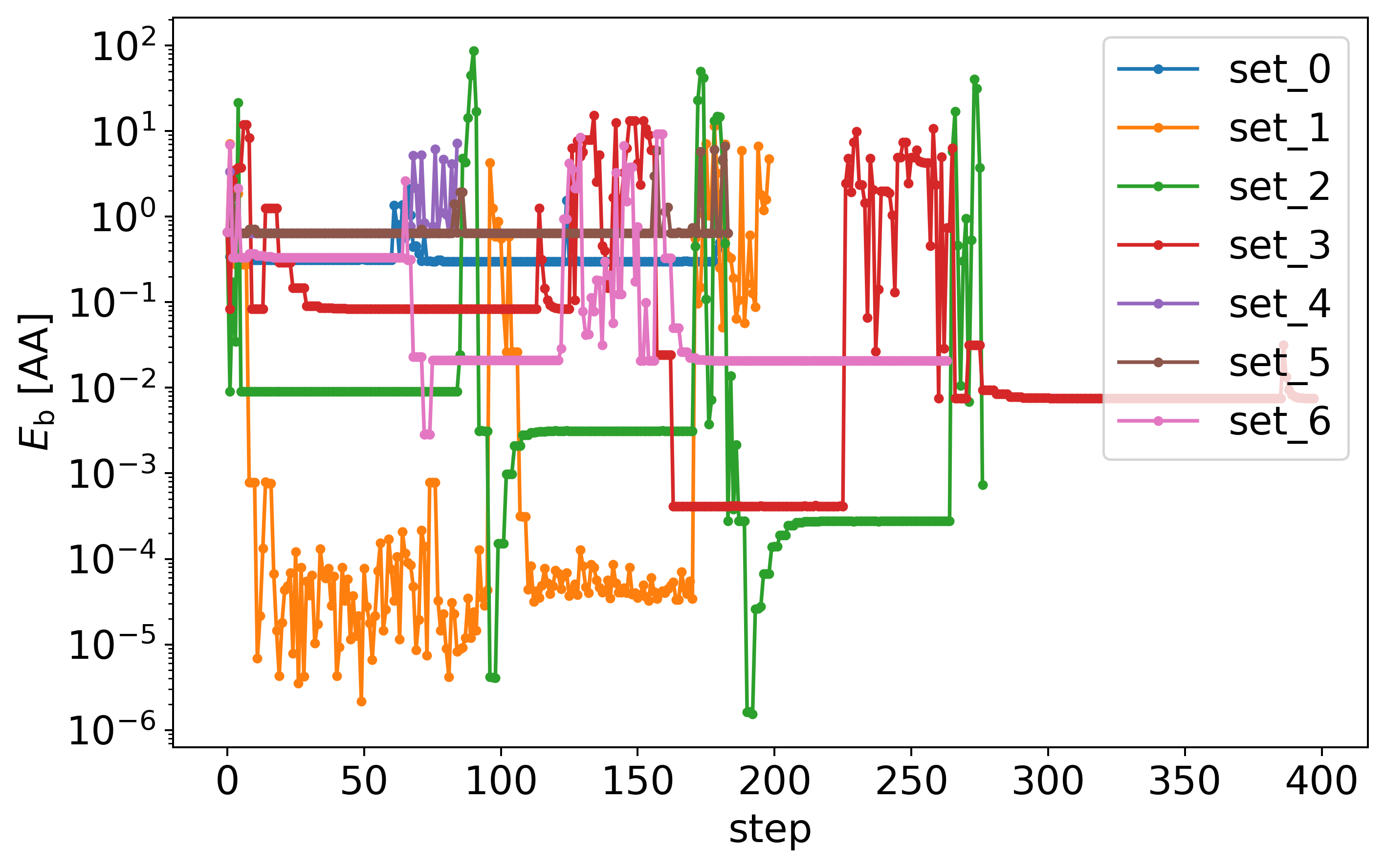
Here are the energies during those parameter optimisations.
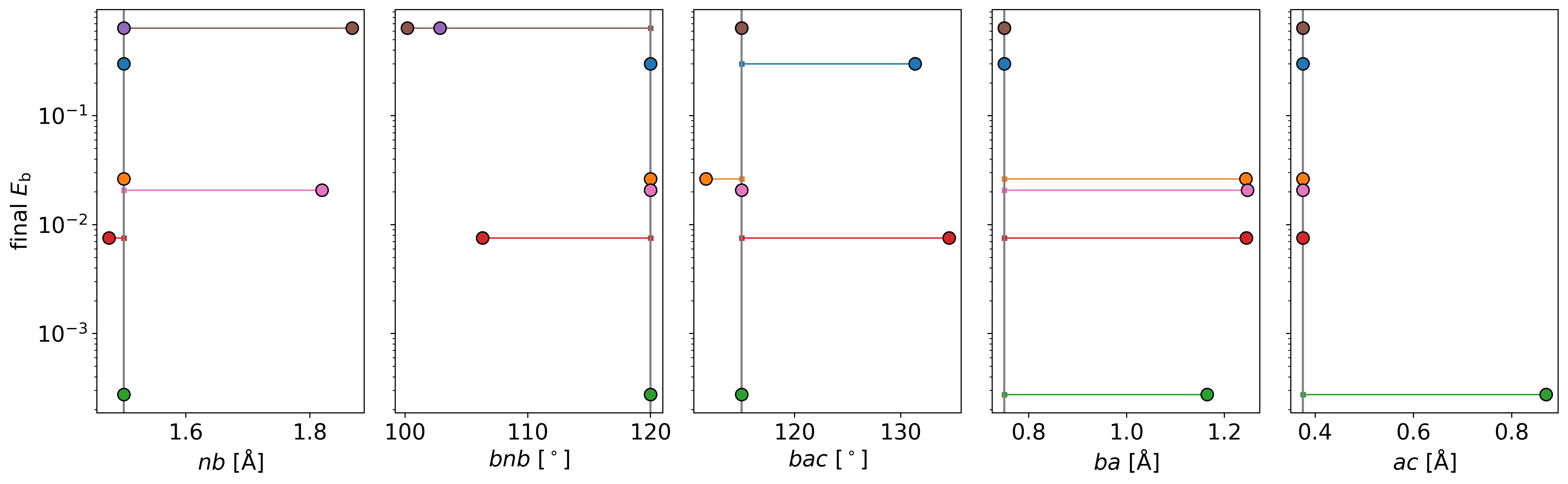
And the changes in the terms for each parameter set. The data here suggests
that stable structures can be achieved by more by changing the ditopic
building block terms (ba, ac and bac). But even small changes bring
th energies very close to 0 kJ/mol.
And a visualisation of the structures:
