Minimal model structure prediction¶
In this recipe, I will reproduce the prediction of the starship structures
from this paper.
Using minimal models of ditopic and tetratopic building blocks, we generated
all possible heteroleptic graphs with various stoichiometries and compute
their relative stability, aiming for the most stable structure. Note that the
forcefield is not modified, but defined from atomistic building blocks.
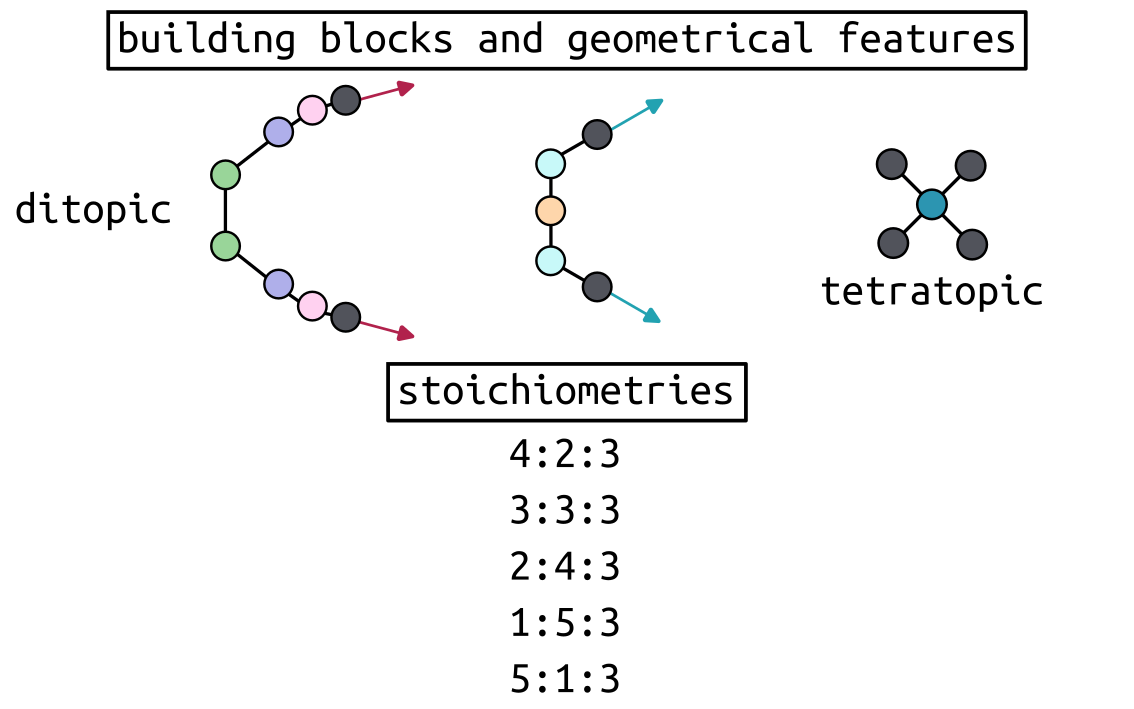
We will start by defining our definer_dict for the constant terms, and the
associated bead library.
# Define a definer dictionary.
# These are constants, while different systems can override these
# parameters.
cg_scale = 2
constant_definer_dict = {
# Bonds.
"mb": ("bond", 1.0, 1e5),
# Angles.
"bmb": ("pyramid", 90, 1e2),
"mba": ("angle", 180, 1e2),
"mbg": ("angle", 180, 1e2),
"aca": ("angle", 180, 1e2),
# Torsions.
"bacab": ("tors", "0134", 180, 50, 1),
"edde": ("tors", "0123", 180.0, 50.0, 1),
"mbge": ("tors", "0123", 180.0, 50.0, 1),
# Nonbondeds.
"m": ("nb", 10.0, 1.0),
"d": ("nb", 10.0, 1.0),
"e": ("nb", 10.0, 1.0),
"a": ("nb", 10.0, 1.0),
"b": ("nb", 10.0, 1.0),
"c": ("nb", 10.0, 1.0),
"g": ("nb", 10.0, 1.0),
}
# Define beads.
bead_library = cgx.molecular.BeadLibrary.from_bead_types(
# Type and coordination.
{"m": 4, "a": 2, "b": 2, "c": 2, "d": 2, "e": 2, "g": 2}
)
Then we can map that to our building block library. In this example, I am just using one pair of ditopic building blocks, unlike in the manuscript.
# Define your forcefield alterations as building blocks.
building_block_library = {
"la": {
"precursor": cgx.molecular.SixBead(
bead=bead_library.get_from_type("d"),
abead1=bead_library.get_from_type("e"),
abead2=bead_library.get_from_type("g"),
),
"mod_definer_dict": {
"dd": ("bond", 7.0 / cg_scale, 1e5),
"de": ("bond", 1.5 / cg_scale, 1e5),
"dde": ("angle", 170, 1e2),
"eg": ("bond", 1.4 / cg_scale, 1e5),
"gb": ("bond", 1.4 / cg_scale, 1e5),
"egb": ("angle", 120, 1e2),
"deg": ("angle", 180, 1e2),
},
},
"st5": {
"precursor": cgx.molecular.TwoC1Arm(
bead=bead_library.get_from_type("c"),
abead1=bead_library.get_from_type("a"),
),
"mod_definer_dict": {
"ba": ("bond", 2.8 / cg_scale, 1e5),
"ac": ("bond", 3.9 / 2 / cg_scale, 1e5),
"bac": ("angle", 120, 1e2),
},
},
"tetra": {
"precursor": cgx.molecular.FourC1Arm(
bead=bead_library.get_from_type("m"),
abead1=bead_library.get_from_type("b"),
),
"mod_definer_dict": {},
},
}
And define a series of systems to explore. Here, I want to check all possible stoichiometry mixtures of these three building blocks, with various multipliers.
# Define systems to predict the structure of.
systems = {
"la_st5_423": {
"stoichiometry_map": {"tetra": 3, "la": 4, "st5": 2},
"multipliers": (1,),
"vdw_cutoff": 2,
},
"la_st5_111": {
"stoichiometry_map": {"tetra": 1, "la": 1, "st5": 1},
"multipliers": (3,),
"vdw_cutoff": 2,
},
"la_st5_243": {
"stoichiometry_map": {"tetra": 3, "la": 2, "st5": 4},
"multipliers": (1,),
"vdw_cutoff": 2,
},
"la_st5_153": {
"stoichiometry_map": {"tetra": 3, "la": 1, "st5": 5},
"multipliers": (1,),
"vdw_cutoff": 2,
},
"la_st5_513": {
"stoichiometry_map": {"tetra": 3, "la": 5, "st5": 1},
"multipliers": (1,),
"vdw_cutoff": 2,
},
}
Time to iterate!
for system_name, syst_d in systems.items():
logger.info("doing system: %s", system_name)
# Merge constant dict with modifications from different systems.
merged_definer_dicts = cgx.systems_optimisation.merge_definer_dicts(
original_definer_dict=constant_definer_dict,
new_definer_dicts=[
building_block_library[i]["mod_definer_dict"]
for i in syst_d["stoichiometry_map"]
],
)
forcefield = cgx.systems_optimisation.get_forcefield_from_dict(
identifier=f"{system_name}ff",
prefix=f"{system_name}ff",
vdw_bond_cutoff=syst_d["vdw_cutoff"],
present_beads=bead_library.get_present_beads(),
definer_dict=merged_definer_dicts,
)
# Build all the building blocks and pre optimise their structures.
bb_map = {}
for prec_name in syst_d["stoichiometry_map"]:
prec = building_block_library[prec_name]["precursor"]
bb = cgx.utilities.optimise_ligand(
molecule=prec.get_building_block(),
name=f"{system_name}_{prec.get_name()}",
output_dir=calc_dir,
forcefield=forcefield,
platform=None,
).clone()
bb.write(
str(ligand_dir / f"{system_name}_{prec.get_name()}_optl.mol")
)
bb_map[prec_name] = bb
for multiplier in syst_d["multipliers"]:
logger.info(
"doing system: %s, multi: %s", system_name, multiplier
)
# Define a connectivity based on a multiplier.
iterator = cgx.scram.TopologyIterator(
building_block_counts={
bb_map[name]: stoich * multiplier
for name, stoich in syst_d["stoichiometry_map"].items()
},
)
logger.info(
"graph iteration has %s graphs", iterator.count_graphs()
)
possible_bbdicts = iterator.get_configurations()
logger.info(
"building block iteration has %s options",
len(possible_bbdicts),
)
logger.info(
"iterating over %s graphs and bb configurations...",
iterator.count_graphs() * len(possible_bbdicts),
)
run_topology_codes: list[agx.ConfiguredCode] = []
for bb_config, topology_code in it.product(
possible_bbdicts,
iterator.yield_graphs(),
):
# Filter graphs for 1-loops.
if topology_code.contains_parallels():
continue
configured = agx.ConfiguredCode(topology_code, bb_config)
if not agx.utilities.is_configured_code_isomorphic(
test_code=configured,
run_topology_codes=run_topology_codes,
):
continue
run_topology_codes.append(configured)
# Here we apply a multi-initial state, multi-step geometry
# optimisation algorithm.
config_name = (
f"{system_name}_{multiplier}_"
f"{topology_code.idx}_b{bb_config.idx}"
)
# Each conformer is stored here.
conformer_db_path = calc_dir / f"{config_name}.db"
optimisation_workflow(
config_name=config_name,
conformer_db_path=conformer_db_path,
topology_code=topology_code,
iterator=iterator,
bb_config=bb_config,
calculation_dir=calc_dir,
forcefield=forcefield,
)
conformer_db = cgx.utilities.AtomliteDatabase(
conformer_db_path
)
min_energy_structure = None
min_energy = float("inf")
min_energy_key = None
for entry in conformer_db.get_entries():
if entry.properties["energy_per_bb"] < min_energy:
min_energy = entry.properties["energy_per_bb"]
min_energy_structure = conformer_db.get_molecule(
key=entry.key
)
min_energy_key = entry.key
# To file.
min_energy_structure.write(
str(struct_output / f"{config_name}_optc.mol")
)
# To database.
cgx.utilities.AtomliteDatabase(database_path).add_molecule(
molecule=min_energy_structure, key=config_name
)
properties = {
"multiplier": multiplier,
"topology_idx": topology_code.idx,
}
cgx.utilities.AtomliteDatabase(database_path).add_properties(
key=config_name, property_dict=properties
)
analyse_cage(
database_path=database_path,
name=config_name,
conformer_db_path=conformer_db_path,
min_energy_key=min_energy_key,
)
And now we can plot the most stable structure for each multiplier to show the star ship is indeed the most stable.
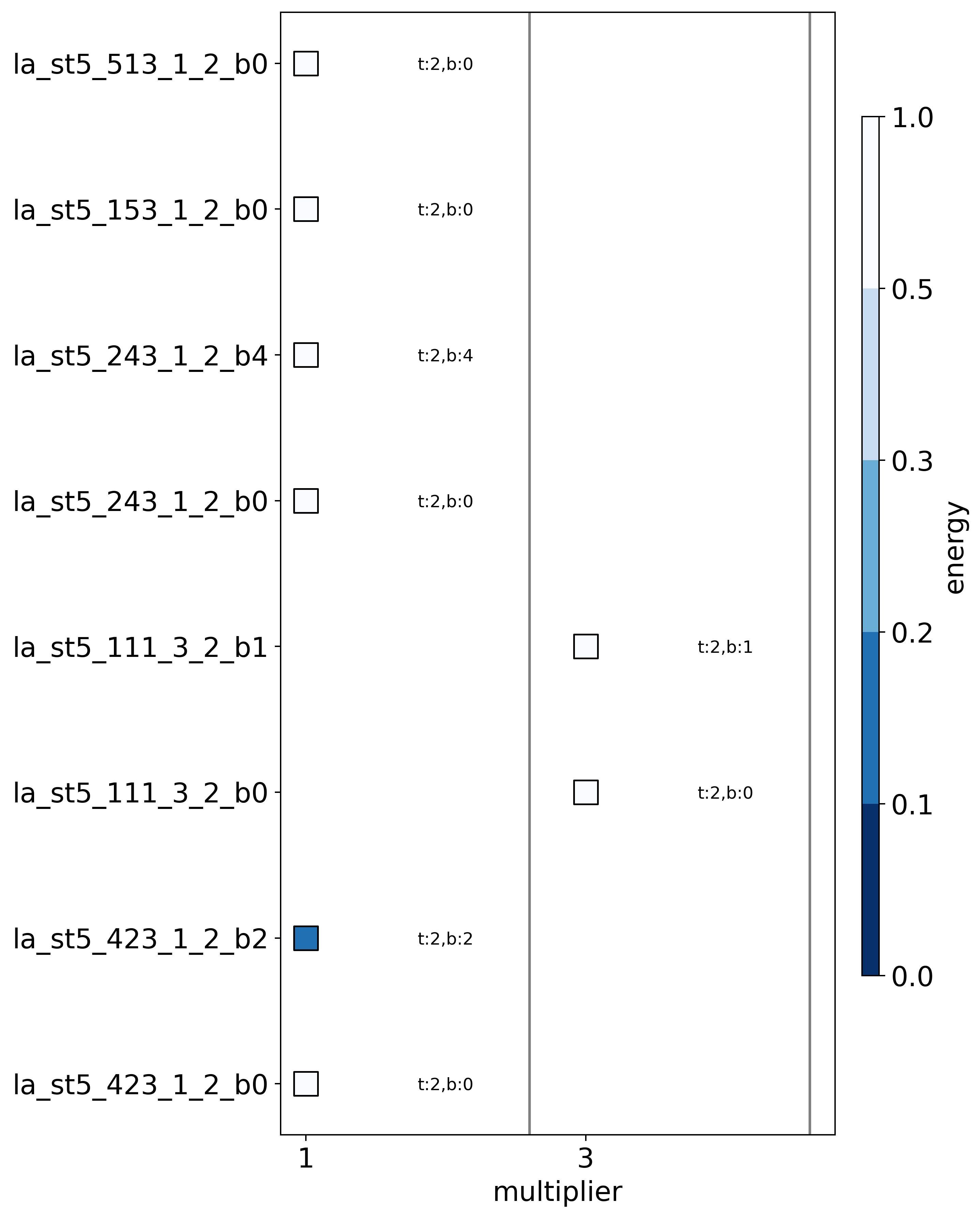
With the structure:
⬇️ Download Python Script