Genetic algorithm-driven graph exploration¶
To simplify this example, which mirrors our recently published example, I am showing the beginnings of exploring graph and building block configurations with a hierarchical genetic algorihm, with the shortest possible geometry optimisation (hence, the results may not be as described in the paper).
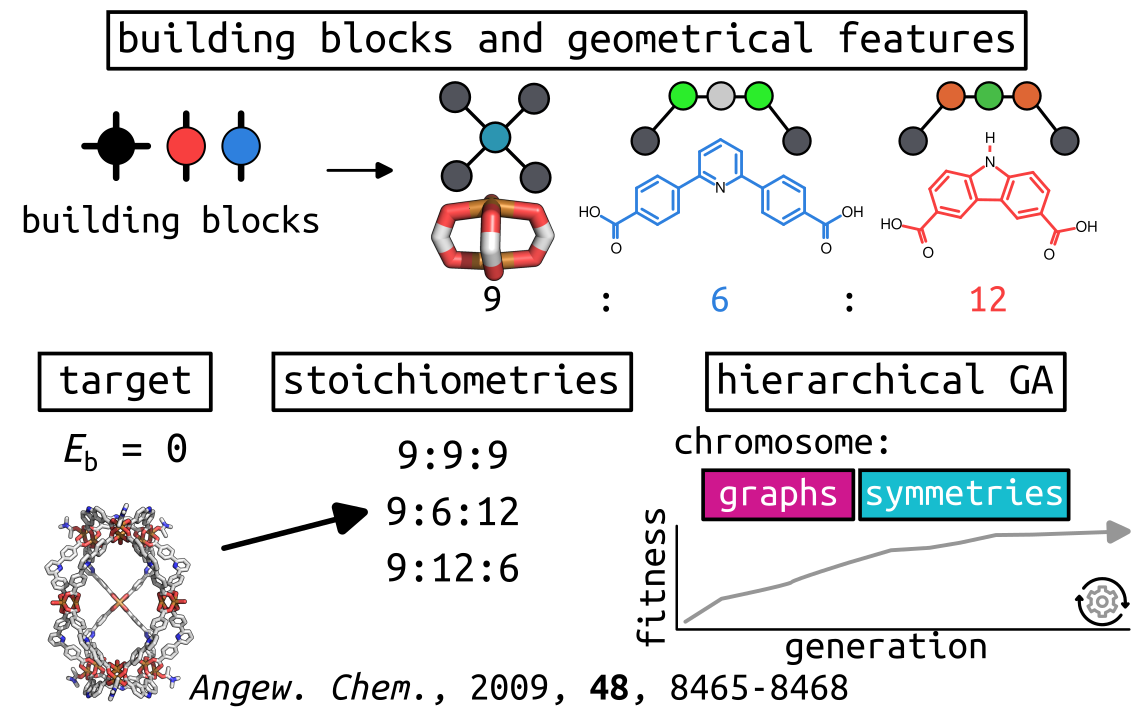
As per usual, we will define our building blocks and systems:
# Define beads.
bead_library = cgx.molecular.BeadLibrary.from_bead_types(
# Type and coordination.
{"m": 4, "a": 2, "e": 2, "b": 2, "c": 2, "d": 2}
)
# Define a definer dictionary.
# These are constants, while different systems can override these
# parameters.
cg_scale = 2
constant_definer_dict = {
# Bonds.
"mb": ("bond", 1.0, 1e5),
# Angles.
"bmb": ("pyramid", 90, 1e2),
"mba": ("angle", 180, 1e2),
"mbe": ("angle", 180, 1e2),
"aca": ("angle", 180, 1e2),
"ede": ("angle", 180, 1e2),
# Torsions.
"bacab": ("tors", "0134", 180, 50, 1),
"bedeb": ("tors", "0134", 180, 50, 1),
# Nonbondeds.
"m": ("nb", 10.0, 1.0),
"d": ("nb", 10.0, 1.0),
"e": ("nb", 10.0, 1.0),
"a": ("nb", 10.0, 1.0),
"b": ("nb", 10.0, 1.0),
"c": ("nb", 10.0, 1.0),
}
# Define your forcefield alterations as building blocks.
building_block_library = {
"deg90": {
"precursor": cgx.molecular.TwoC1Arm(
bead=bead_library.get_from_type("d"),
abead1=bead_library.get_from_type("e"),
),
"mod_definer_dict": {
"be": ("bond", 1.5 / cg_scale, 1e5),
"de": ("bond", 5.9 / 2 / cg_scale, 1e5),
"deb": ("angle", 135, 1e2),
},
},
"l1a": {
"precursor": cgx.molecular.TwoC1Arm(
bead=bead_library.get_from_type("c"),
abead1=bead_library.get_from_type("a"),
),
"mod_definer_dict": {
"ba": ("bond", 1.5 / cg_scale, 1e5),
"ac": ("bond", 9.5 / 2 / cg_scale, 1e5),
"bac": ("angle", 150, 1e2),
},
},
"tetra": {
"precursor": cgx.molecular.FourC1Arm(
bead=bead_library.get_from_type("m"),
abead1=bead_library.get_from_type("b"),
),
"mod_definer_dict": {},
},
}
# Define systems to predict the structure of.
# Only focussing on m=9.
multiplier = 1
systems = {
"l1a_90_6-12-9": {
"stoichiometry_map": {"tetra": 9, "l1a": 6, "deg90": 12},
"vdw_cutoff": 2,
},
"l1a_90_12-6-9": {
"stoichiometry_map": {"tetra": 9, "l1a": 12, "deg90": 6},
"vdw_cutoff": 2,
},
"l1a_90_9-9-9": {
"stoichiometry_map": {"tetra": 9, "l1a": 9, "deg90": 9},
"vdw_cutoff": 2,
},
}
I will not show the iteration in detail, because it is quite long, but here are some functions we will use.
Firstly, the structure and fitness functions:
def fitness_function( # noqa: PLR0913
chromosome: cgx.systems_optimisation.Chromosome,
chromosome_generator: cgx.systems_optimisation.ChromosomeGenerator, # noqa: ARG001
database_path: pathlib.Path,
calculation_output: pathlib.Path, # noqa: ARG001
structure_output: pathlib.Path, # noqa: ARG001
options: dict,
) -> float:
"""Calculate fitness."""
database = cgx.utilities.AtomliteDatabase(database_path)
topology_idx, _ = chromosome.get_topology_information()
building_block_config = chromosome.get_building_block_configurations()[0]
name = f"{chromosome.prefix}_{topology_idx}_b{building_block_config.idx}"
entry = database.get_entry(name)
if not entry.properties["opt_passed"]:
energy = 100
elif entry.properties["is_duplicate"]:
energy = database.get_entry(
entry.properties["duplicate_of"]
).properties["energy_per_bb"]
else:
energy = entry.properties["energy_per_bb"]
fitness = np.exp(-energy * options["beta"])
database.add_properties(key=name, property_dict={"fitness": fitness})
return fitness
Importantly, the structure function will check for duplicate graphs and configurations to avoid rerunning calculations.
def structure_function( # noqa: C901, PLR0915
chromosome: cgx.systems_optimisation.Chromosome,
database_path: pathlib.Path,
calculation_output: pathlib.Path,
structure_output: pathlib.Path,
options: dict,
) -> None:
"""Geometry optimisation."""
database = cgx.utilities.AtomliteDatabase(database_path)
topology_idx, topology_code = chromosome.get_topology_information()
building_block_config = chromosome.get_building_block_configurations()[0]
base_name = (
f"{chromosome.prefix}_{topology_idx}_b{building_block_config.idx}"
)
l1, l2, stoichstring = chromosome.prefix.split("_")
multiplier = stoichstring.split("-")[2]
if database.has_molecule(base_name):
return
# Check if this has been run before.
known_entry = None
for entry in database.get_entries():
# Only do base entries.
if "is_base" not in entry.properties:
continue
try:
entry_tc = options["topology_codes"][
entry.properties["topology_idx"]
]
entry_bb_config = options["bb_configs"][
entry.properties["bb_config_idx"]
]
except (KeyError, IndexError):
continue
known_stoichstring = entry.properties["stoichstring"]
known_pair = entry.properties["pair"]
if f"{l1}_{l2}" != known_pair:
continue
if stoichstring != known_stoichstring:
continue
# Testing bb-config aware graph check.
configured = agx.ConfiguredCode(topology_code, building_block_config)
if not agx.utilities.is_configured_code_isomorphic(
test_code=configured,
run_topology_codes=[agx.ConfiguredCode(entry_tc, entry_bb_config)],
):
known_entry = entry
break
# Try to avoid recalculation if possible.
if (
known_entry is not None
and known_entry.properties["base_name"] != base_name
):
database.add_molecule(
key=base_name,
molecule=database.get_molecule(known_entry.key),
)
database.add_properties(
key=base_name,
property_dict={
"is_duplicate": True,
"duplicate_of": known_entry.key,
"opt_passed": True,
},
)
try:
nd_ = known_entry.properties["num_duplicates"] + 1
except KeyError:
nd_ = 1
database.add_properties(
key=known_entry.key,
property_dict={"num_duplicates": nd_},
)
logger.info("%s is duplicate", base_name)
return
# Actually do the calculation, now, just because we have too.
constructed_molecule = cgx.scram.get_regraphed_molecule(
layout_type="kamada",
scale=10,
topology_code=topology_code,
iterator=options["iterator"],
configuration=building_block_config,
)
constructed_molecule.write(calculation_output / f"{base_name}_unopt.mol")
opt_file = structure_output / f"{base_name}_optc.mol"
# Optimise and save.
logger.info("building %s", base_name)
try:
conformer = cgx.utilities.run_optimisation(
assigned_system=options["forcefield"].assign_terms(
molecule=constructed_molecule,
name=base_name,
output_dir=calculation_output,
),
name=base_name,
file_suffix="opt1",
output_dir=calculation_output,
platform=None,
)
opt_passed = True
except OpenMMException:
logger.info("failed optimisation of %s", base_name)
opt_passed = False
if opt_passed:
properties = {
"base_name": base_name,
"energy_per_bb": cgx.utilities.get_energy_per_bb(
energy_decomposition=conformer.energy_decomposition,
number_building_blocks=(
options["iterator"].get_num_building_blocks()
),
),
"num_components": len(
stko.Network.init_from_molecule(
conformer.molecule
).get_connected_components()
),
"forcefield_dict": (
options["forcefield"].get_forcefield_dictionary()
),
"l1": l1,
"l2": l2,
"pair": f"{l1}_{l2}",
"num_bbs": (options["iterator"].get_num_building_blocks()),
"stoichstring": stoichstring,
"multiplier": multiplier,
"topology_idx": topology_idx,
"topology_code_vmap": tuple(
(int(i[0]), int(i[1])) for i in topology_code.vertex_map
),
"bb_config_idx": building_block_config.idx,
# Add here, if it gets here, then it is not duplicate.
"is_duplicate": False,
"num_duplicates": 0,
}
database.add_molecule(key=base_name, molecule=conformer.molecule)
conformer.molecule.write(opt_file)
else:
database.add_molecule(key=base_name, molecule=constructed_molecule)
# Write base name to database.
database.add_properties(key=base_name, property_dict=properties)
database.add_properties(
key=base_name,
property_dict={"is_base": True, "opt_passed": opt_passed},
)
Then, we can define a single genetic algorithm run in this function, where we use random and roulette selection for mutations and crossovers over the graph and building block configurations:
Note
The building block configurations are added to the chromosome generator
as building_block_configurations, while the graphs are added as
topology. Hence, they are accessed through get_bc_ids() and
get_topo_ids(), respectively.
def run_genetic_algorithm( # noqa: PLR0913
seed: int,
chromo_it: cgx.systems_optimisation.ChromosomeGenerator,
fitness_calculator: cgx.systems_optimisation.FitnessCalculator,
structure_calculator: cgx.systems_optimisation.StructureCalculator,
scan_config: dict,
current_population: cgx.systems_optimisation.Generation | None,
database: cgx.utilities.AtomliteDatabase,
neighbour_opt: bool,
) -> list[float]:
"""A helper function for running each GA."""
generator = np.random.default_rng(seed)
if current_population is None:
initial_population = chromo_it.select_random_population(
generator,
size=scan_config["selection_size"],
)
else:
logger.info(
"selected elite with f>%s",
round(
current_population.calculate_elite_fitness(
proportion_threshold=0.25
),
5,
),
)
initial_population = current_population.select_elite(
proportion_threshold=0.25
)
# Yield this.
generations = []
generation = cgx.systems_optimisation.Generation(
chromosomes=initial_population,
fitness_calculator=fitness_calculator,
structure_calculator=structure_calculator,
num_processes=scan_config["num_processes"],
)
generation.run_structures()
_ = generation.calculate_fitness_values()
generations.append(generation)
for generation_id in range(1, scan_config["num_generations"] + 1):
logger.info("doing generation %s of seed %s", generation_id, seed)
logger.info("initial size is %s.", generation.get_generation_size())
logger.info("doing mutations.")
if neighbour_opt:
merged_chromosomes = []
merged_chromosomes.extend(
chromo_it.get_population_neighbours(
chromosomes={
(
f"{chromosome.prefix}"
f"_{chromosome.get_topology_information()[0]}"
f"_b{chromosome.get_building_block_configurations()[0].idx}"
): chromosome
for chromosome in generation.chromosomes
},
selection="all",
gene_range=chromo_it.get_bc_ids(),
)
)
merged_chromosomes.extend(generation.select_all())
else:
merged_chromosomes = []
merged_chromosomes.extend(
chromo_it.mutate_population(
chromosomes={
(
f"{chromosome.prefix}"
f"_{chromosome.get_topology_information()[0]}"
f"_b{chromosome.get_building_block_configurations()[0].idx}"
): chromosome
for chromosome in generation.chromosomes
},
generator=generator,
gene_range=chromo_it.get_bc_ids(),
selection="random",
num_to_select=scan_config["mutations"],
database=database,
)
)
merged_chromosomes.extend(
chromo_it.mutate_population(
chromosomes={
(
f"{chromosome.prefix}"
f"_{chromosome.get_topology_information()[0]}"
f"_b{chromosome.get_building_block_configurations()[0].idx}"
): chromosome
for chromosome in generation.chromosomes
},
generator=generator,
gene_range=chromo_it.get_topo_ids(),
selection="random",
num_to_select=scan_config["mutations"],
database=database,
)
)
merged_chromosomes.extend(
chromo_it.mutate_population(
chromosomes={
(
f"{chromosome.prefix}"
f"_{chromosome.get_topology_information()[0]}"
f"_b{chromosome.get_building_block_configurations()[0].idx}"
): chromosome
for chromosome in generation.chromosomes
},
generator=generator,
gene_range=chromo_it.get_bc_ids(),
selection="roulette",
num_to_select=scan_config["mutations"],
database=database,
)
)
merged_chromosomes.extend(
chromo_it.mutate_population(
chromosomes={
(
f"{chromosome.prefix}"
f"_{chromosome.get_topology_information()[0]}"
f"_b{chromosome.get_building_block_configurations()[0].idx}"
): chromosome
for chromosome in generation.chromosomes
},
generator=generator,
gene_range=chromo_it.get_topo_ids(),
selection="roulette",
num_to_select=scan_config["mutations"],
database=database,
)
)
merged_chromosomes.extend(
chromo_it.crossover_population(
chromosomes={
(
f"{chromosome.prefix}"
f"_{chromosome.get_topology_information()[0]}"
f"_b{chromosome.get_building_block_configurations()[0].idx}"
): chromosome
for chromosome in generation.chromosomes
},
generator=generator,
selection="random",
num_to_select=scan_config["mutations"],
database=database,
)
)
merged_chromosomes.extend(
chromo_it.crossover_population(
chromosomes={
(
f"{chromosome.prefix}"
f"_{chromosome.get_topology_information()[0]}"
f"_b{chromosome.get_building_block_configurations()[0].idx}"
): chromosome
for chromosome in generation.chromosomes
},
generator=generator,
selection="roulette",
num_to_select=scan_config["mutations"],
database=database,
)
)
# Add the best 5 to the new generation.
merged_chromosomes.extend(generation.select_best(selection_size=5))
generation = cgx.systems_optimisation.Generation(
chromosomes=chromo_it.dedupe_population(merged_chromosomes),
fitness_calculator=fitness_calculator,
structure_calculator=structure_calculator,
num_processes=scan_config["num_processes"],
)
logger.info("new size is %s.", generation.get_generation_size())
# Build, optimise and analyse each structure.
generation.run_structures()
_ = generation.calculate_fitness_values()
# Add final state to generations.
generations.append(generation)
# Select the best of the generation for the next
# generation.
best = generation.select_best(
selection_size=scan_config["selection_size"]
)
generation = cgx.systems_optimisation.Generation(
chromosomes=chromo_it.dedupe_population(best),
fitness_calculator=fitness_calculator,
structure_calculator=structure_calculator,
num_processes=scan_config["num_processes"],
)
logger.info("final size is %s.", generation.get_generation_size())
# Output best structures as images.
best_chromosome = generation.select_best(selection_size=1)[0]
best_name = (
f"{best_chromosome.prefix}_"
f"{best_chromosome.get_topology_information()[0]}_"
f"b{best_chromosome.get_building_block_configurations()[0].idx}"
)
logger.info("top scorer is %s (seed: %s)", best_name, seed)
return generations
Now, the hierarchical genetic algorithm runs look like these calls to the
run_genetic_algorithm function. Note that we limit the search space
siginficantly for the sake of the test here.
seeded_generations = {}
# Very short runs!
scan_config = {
"seeds": [4, 12689],
"mutations": 2,
"num_generations": 5,
"selection_size": 5,
"num_processes": 1,
"long_seeds": [142],
"neighbour_seeds": [6582],
}
for seed in scan_config["seeds"]:
seeded_generations[seed] = run_genetic_algorithm(
seed=seed,
chromo_it=chromo_it,
fitness_calculator=fitness_calculator,
structure_calculator=structure_calculator,
scan_config=scan_config,
database=database,
current_population=None,
neighbour_opt=False,
)
progress_plot(
seeded_generations=seeded_generations,
output=figure_dir / f"fp_{system_name}.png",
)
# Run longer GA from elites.
chromosomes = []
for generations in seeded_generations.values():
for generation in generations:
chromosomes.extend(generation.chromosomes)
current_population = cgx.systems_optimisation.Generation(
chromosomes=chromo_it.dedupe_population(chromosomes),
fitness_calculator=fitness_calculator,
structure_calculator=structure_calculator,
num_processes=scan_config["num_processes"], # type:ignore[arg-type]
)
for seed in scan_config["long_seeds"]:
temp_scan_config = scan_config.copy()
temp_scan_config.update(
{"num_generations": scan_config["num_generations"] * 2}
)
seeded_generations[seed] = run_genetic_algorithm(
seed=seed,
chromo_it=chromo_it,
fitness_calculator=fitness_calculator,
structure_calculator=structure_calculator,
scan_config=temp_scan_config,
database=database,
current_population=current_population,
neighbour_opt=False,
)
progress_plot(
seeded_generations=seeded_generations,
output=figure_dir / f"fp_{system_name}.png",
)
# And then again, but only over neighbours.
chromosomes = []
for generations in seeded_generations.values():
for generation in generations:
chromosomes.extend(generation.chromosomes)
current_population = cgx.systems_optimisation.Generation(
chromosomes=chromo_it.dedupe_population(chromosomes),
fitness_calculator=fitness_calculator,
structure_calculator=structure_calculator,
num_processes=scan_config["num_processes"], # type:ignore[arg-type]
)
for seed in scan_config["neighbour_seeds"]:
temp_scan_config = scan_config.copy()
temp_scan_config.update({"selection_size": 50})
temp_scan_config.update(
{"num_generations": scan_config["num_generations"] * 2}
)
seeded_generations[seed] = run_genetic_algorithm(
seed=seed,
chromo_it=chromo_it,
fitness_calculator=fitness_calculator,
structure_calculator=structure_calculator,
scan_config=temp_scan_config,
database=database,
current_population=current_population,
neighbour_opt=True,
)
progress_plot(
seeded_generations=seeded_generations,
output=figure_dir / f"fp_{system_name}.png",
)
This process (for one stoichiometry) generated 612 structures
out of the possible 297024 (not considering duplicate symmetries)!
However, with the cheap optimisation process, it did not reproduce the
experimental outcome, where the s=12:6:9 system is the most stable. This
highlights the importance of the geometry optimisation workflow.
Indeed the energy distribution shows that the lowest energy structure has the
wrong stoichiometry.
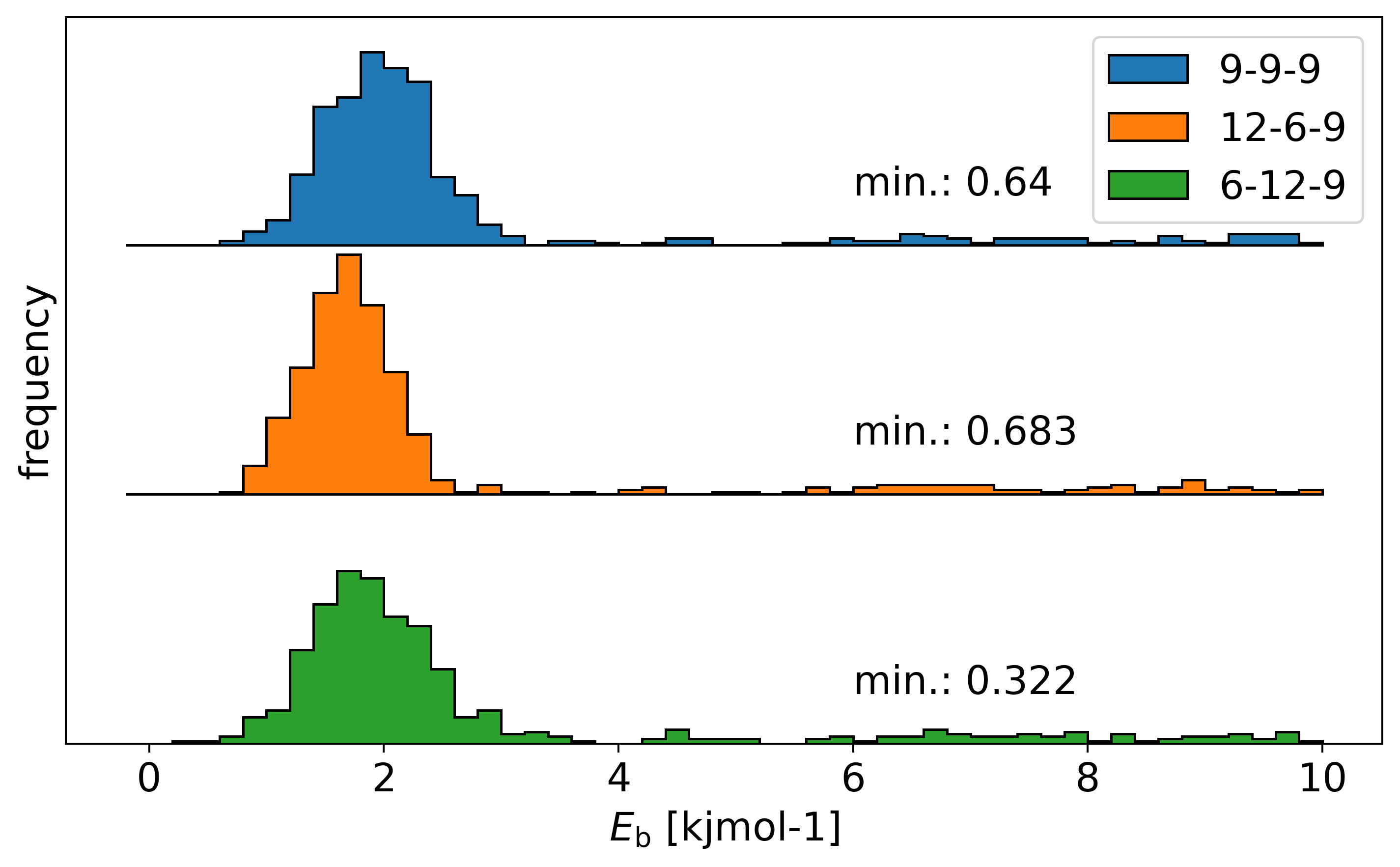
And here is the lowest energy structure of all stoiochiometries:
Which does not look like the most stable structure from the manuscript:
Here are the three fitness plots for the three tested stoichiometries:
s=9:9:9:
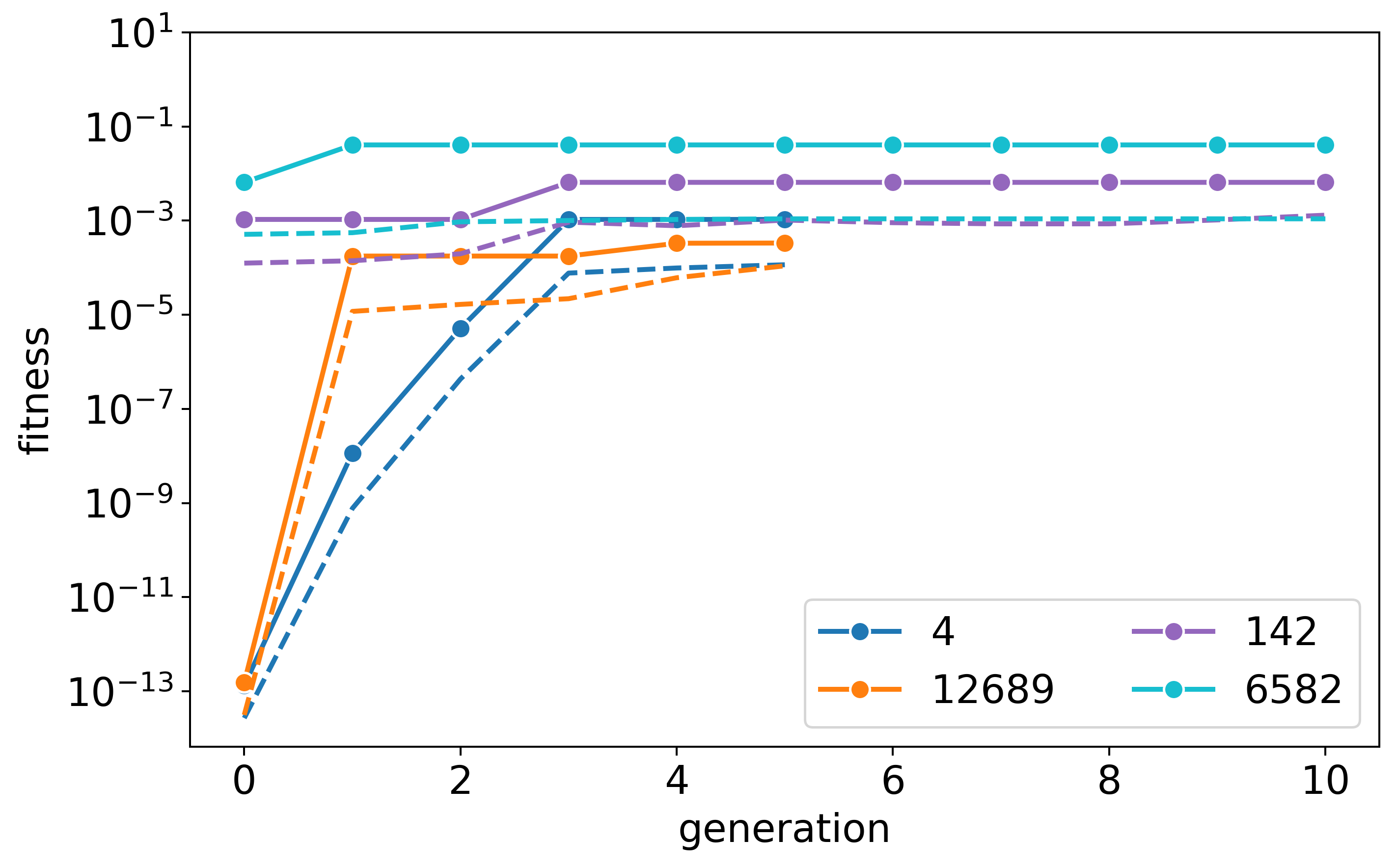
s=6:12:9:
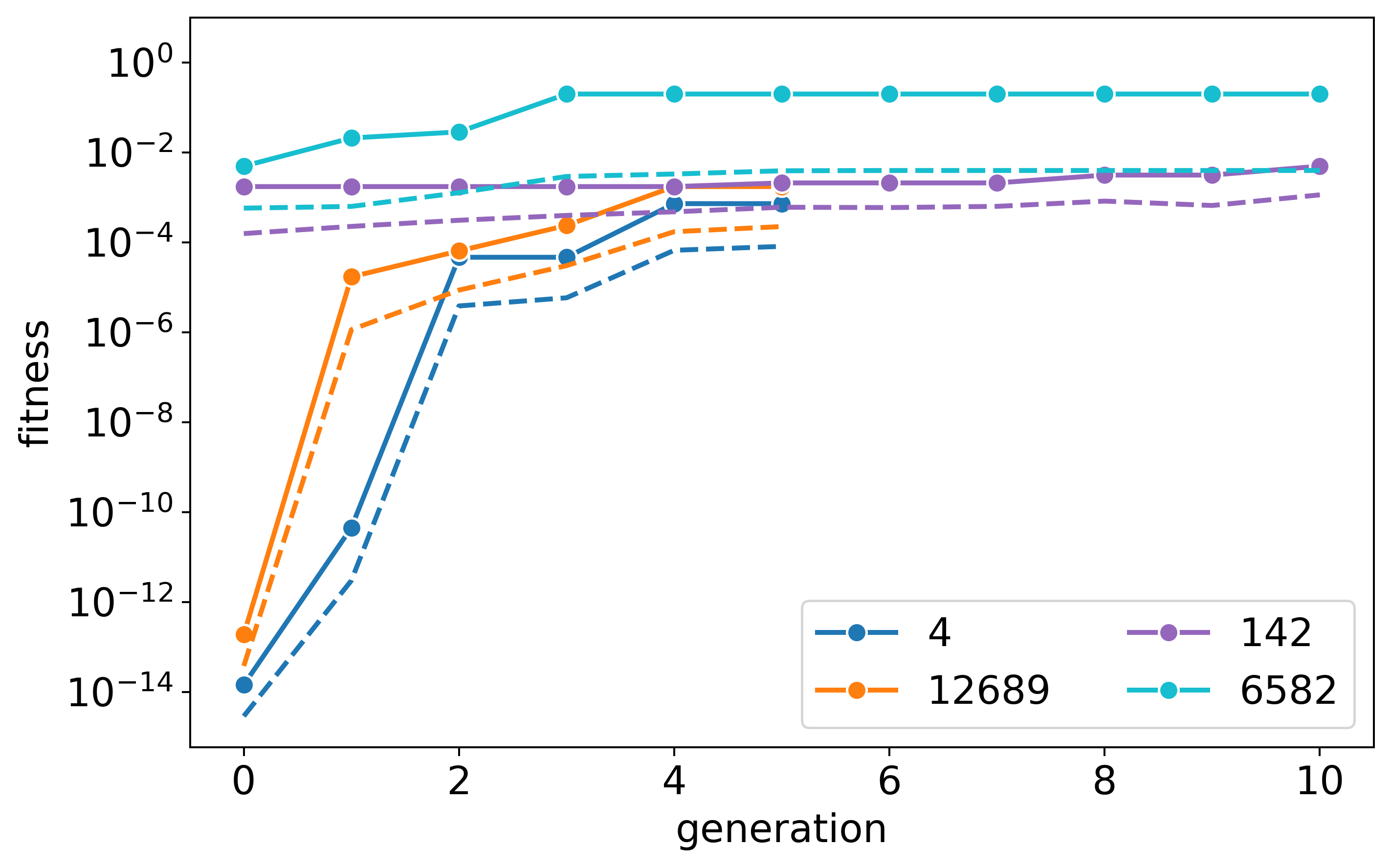
s=12:6:9:
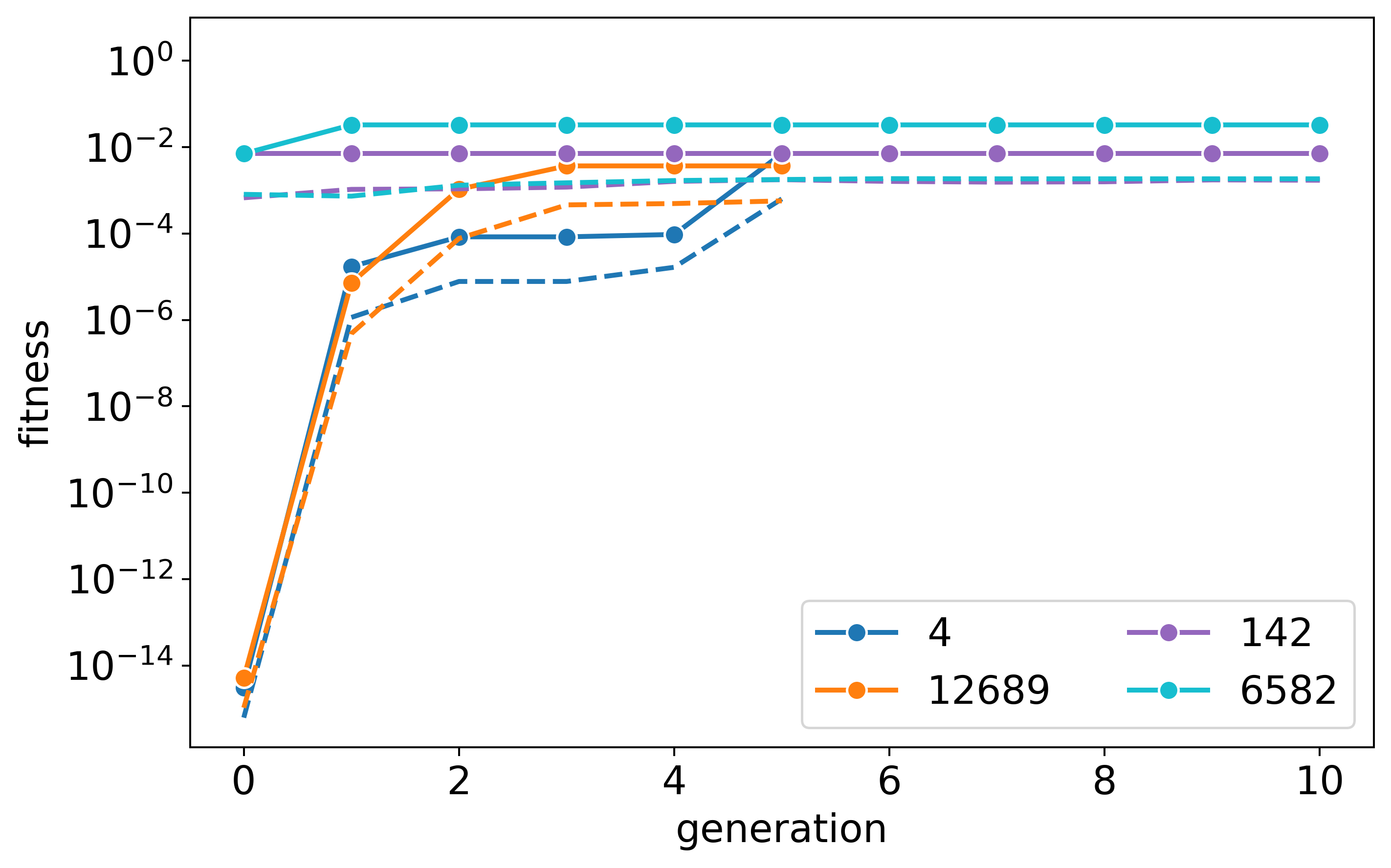
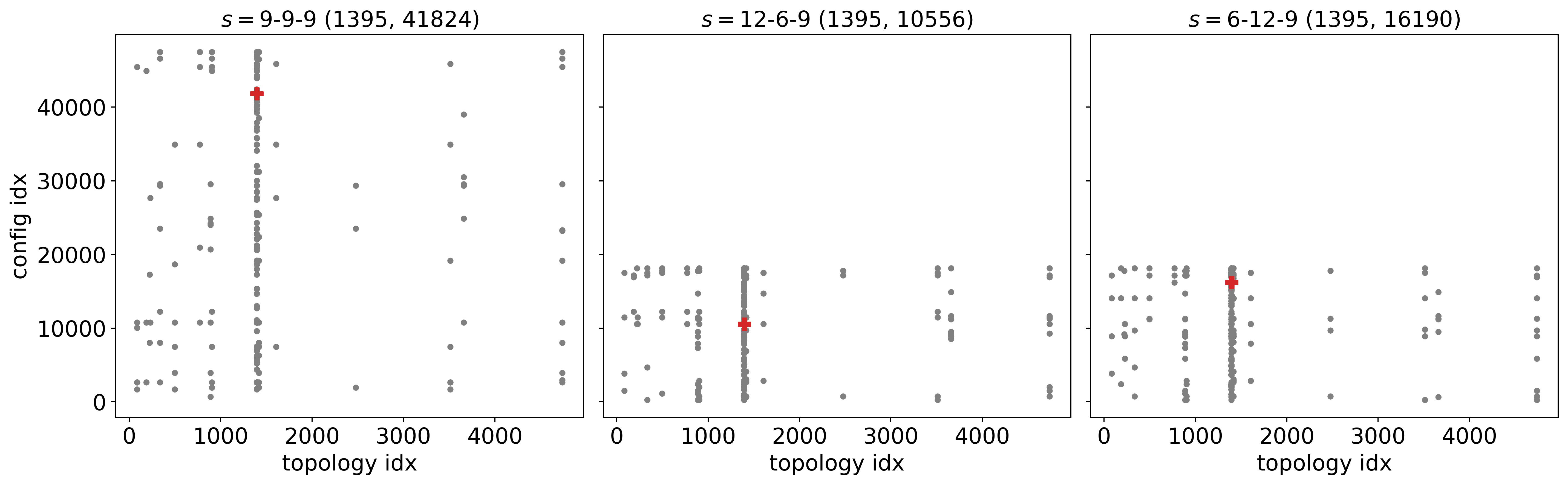
An example of the graph and building block configuration exploration:
 ⬇️ Download Python Script
⬇️ Download Python Script